

Transmission electron microscope image of negative-stained, Fortaleza-strain Zika virus (red), isolated from a microcephaly case in Brazil. Credit: NIAID.
Researchers have found that Zika virus (ZIKV), the mosquito-borne virus which is associated with ZIKV congenital syndrome, may have been present in many affected regions months before it was first detected.
What is Zika?
ZIKV is a flavivirus which is transmitted primarily through the Aedes mosquito. Infection with ZIKV causes similar symptoms to the tropical disease Dengue fever such as rash, skeletomuscular pain and fever. These symptoms are usually mild and last 2 – 7 days. However, ZIKA has more serious implications if contracted in pregnancy, as it can cause congenital brain abnormalities, such as microcephaly [1]. The unprecedented outbreak of ZIKV in the world’s tropical belt has led to a rapid escalation of research into the pathogenesis and transmission of ZIKV.
Why do we need genetic information about Zika?
A consortium of researchers funded in part by the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH), has published a new study in the journal Nature that investigated ZIKV phylogenetic tree (scroll down for a description) and determined if there are any mutations in the viral genetic code. This information is essential to fully understand the trajectory of the outbreak and if there are any new characteristics of the virus which may make it more pathogenic. Additionally, this also helps determine whether current diagnostic tests are still effective at detecting ZIKV in at risk populations [2][4].
Despite the fact that ZIKA was discovered 70 years ago, fewer than 100 ZIKV genomes have been sequenced. This is predominantly due to improper processing of clinical samples and low amounts of virus present in the plasma of infected individuals (significantly lower than other viruses such as Ebola)[2].
Genetic sequencing predicts outbreak origins and mutations
Using human and mosquito samples, the investigators of this study used multiple strategies to create a genetic dataset of 110 ZIKV genomes. Together with 64 published genomes from the scientific literature, they generated a phylogenetic tree and analysed the patterns and timings of when the virus was introduced into geographical regions within the Americas.
The analysis indicates that ZIKV originated in Brazil and rapidly spread to neighboring countries. The authors commented that the speed of transmission early in the outbreak, was probably due to the naivety of the South American population to the virus; meaning their immune systems had not become resistant to ZIKV. The virus diversified into 4 genetically distinct clades or branches of the phylogentic tree (a common ancestor and all its descendants), which spread to Puerto Rico, Honduras, Colombia and the fourth clade included certain Caribbean islands and the continental United States [2].
The researchers also estimate that ZIKV arrived and circulated in certain areas much earlier than the first confirmed case; approximately 4.5 months earlier in Puerto Rico and 9 months earlier in the Caribbean-US areas [2].
Just like humans, small mutations in the viral genetic code can occur over time. Viral mutations can facilitate drug resistance or help the virus to escape the host immune system. 1,030 mutations were detected in the dataset across the whole genome. While the researchers commented that further investigation would be needed to fully understand the functional implications of these mutations, information about ZIKVs genetic variability will provide vital insight into its biology and pathogenesis. Moreover, this knowledge will also no doubt be a valuable resource for the entire ZIKV research community.
In addition, these mutations can render diagnostic tests obsolete. Fortunately, the mutations detected in the genomes are not predicted to affect the performance of the current diagnostic tests. However, the authors warned, that as viral mutation is an ongoing process, continuous monitoring is recommended to ensure there is suitable health surveillance in vulnerable populations [2].
This work is the first to provide significant insight into the evolution and genetic changes in the ZIKV throughout the recent outbreak. This study highlights the importance of appropriate clinical sample handling and provides a strategy to obtain genetic information from viruses with low plasma levels. Details about genomic evolution will help to inform authorities and healthcare workers about best practice for disease monitoring and diagnosis. Furthermore, understanding the intricacies of viral spread can support public policy decisions and public health strategies.
Terms and definitions:
What is a phylogenetic tree?
A phylogenetic tree is a diagram that describes the diversification of a group of organisms through time. The main branch represents a common ancestral lineage and the branches are the descendants of that lineage. Following an event which causes the common ancestor to split or change (speciation), a daughter lineage is created [3][4].
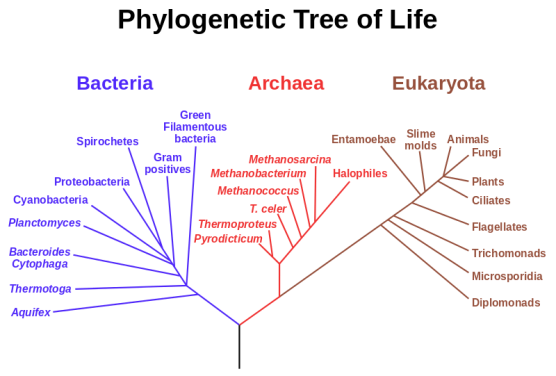
Eric Gaba, NASA Astrobiology Institute
What is a lineage?
A succession of species, where each member has evolved from its predecessor [3][4].
References:
[1] http://www.who.int/mediacentre/factsheets/zika/en/
[2] HC Metsky et al (2017). Genome sequencing reveals Zika virus diversity and spread in the Americas. Nature. 0028-0836. https://www.nature.com/nature/journal/vaop/ncurrent/full/nature22402.html
[3] Larson, Allan, et al (2014). Concepts in Character Macroevolution: Adaptation, Homology, and Evolvability.The Princeton Guide to Evolution, edited by Jonathan B. Losos et al., Princeton University Press, pp. 89–99. http://www.jstor.org/stable/j.ctt4cgc5m
[4] Baum, D. (2008). Reading a Phylogenetic Tree: The Meaning of Monophyletic Groups. Nature Education 1(1):190. https://www.nature.com/scitable/topicpage/reading-a-phylogenetic-tree-the-meaning-of-41956

interesting thanks Erica!
LikeLiked by 1 person
Glad you liked it!
LikeLike